
Research Unit Molecular Cell Biology
and Transgenic Research

Prof. Dr. Paul Saftig
+49 431 880-2216
Prof. Saftig’s work bridges fundamental cell biology (lysosomal trafficking, proteolysis, autophagy) with translational insights in neurodegeneration, lysosomal storage disorders, infectious disease, and cancer. His multidisciplinary contributions — spanning structural biology to in vivo therapies — have significantly advanced our understanding of lysosomal (dys)function and protease networks.
Alzheimer’s Disease & Amyloid Precursor Protein (APP)
- As co-author on seminal studies, he demonstrated that presenilin-1 deficiency affects APP processing—a key mechanism in Alzheimer Disease.
- His research contributed to uncovering Notch signaling via gamma-secretase–like activity
- Honored with the Hans & Ilse Breuer Alzheimer Research Award in 2010 for his contributions to elucidating molecular mechanisms behind the disease.
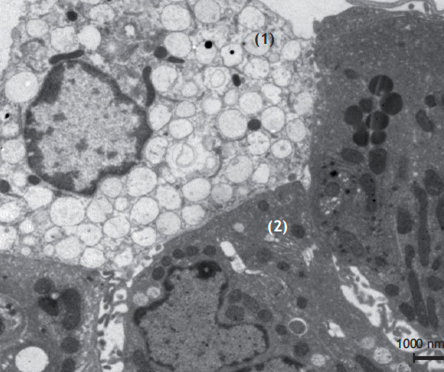
Lysosomal storage in macrophages (taken from J. Cell Sci. 1999, Marquez & Saftig)
Research in Saftig Lab
Viral Entry Mechanisms
- Contribution in a study elucidating how Lassa virus enters host cells via a receptor-switching mechanism, linking lysosomal function to viral infection.
Lysosomal Biology & Lipid Transport
- Pivotal discoveries on LIMP‑2/SCARB2, including its structure and role as a lysosomal cholesterol/lipid exporter and part of ER/lysosome membrane contact sites
- LIMP‑2/SCARB2 was also identified by us as a unique receptor/chaperone for the mannose 6-phosphate-independent transport of the lysosomal hydrolase beta-glucocerebrosidase.
- Our group also explored related abundant lysosomal membrane proteins like LAMP-1/LAMP‑2 and LIMP‑1/CD63, investigating their trafficking and biological functions in autophagy, lysosomal exocytosis, extracellular vesicles and lysosomal biogenesis.
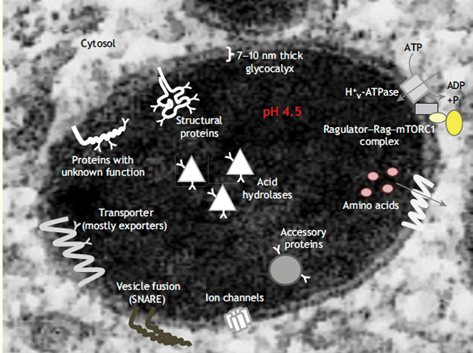
The lysosomal compartment (taken from J. Cell Sci. 1999, Marquez & Saftig)
Therapy of lysosomal storage diseases
- Development of an enzyme replacement therapy (ERT) for alpha‑Mannosidosis, a rare and often fatal lysosomal storage disorder.
- Use and development of the full translational pipeline—from basic science and animal studies to clinical evaluation. Major EU-funded initiatives were initiated, contributed to enzyme production and animal efficacy data, development of clinical trial protocols, and demonstration of safety and preliminary efficacy of ERT in human patients. Our efforts were instrumental in advancing rhLAMAN from concept to a clinically approved therapeutic drug.
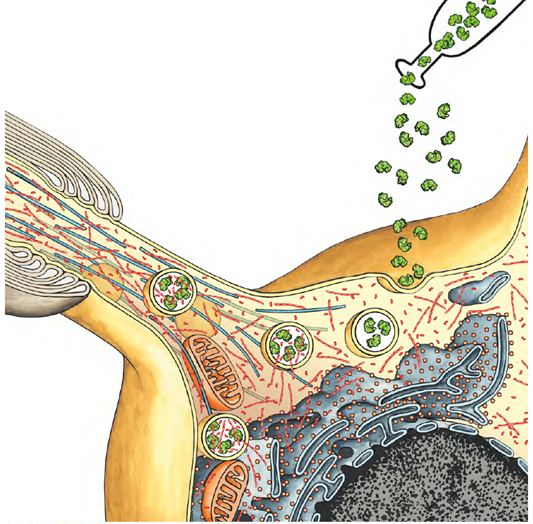
Enzyme Replacement in Lysosome-associated diseases (cover image from Autophagy 2022; Huarcaya et al.)
Proteases & Disease
- Development of a Cathepsin D-based replacement therapy to address proteolysis defects in neuronal ceroid lipofuscinosis (a lysosomal storage disease) and Parkinson Disease models..
- A long-standing focus of the research in the lab is ADAM10, a metalloprotease involved in shedding membrane proteins across several tissues including the kidney’s podocytes.
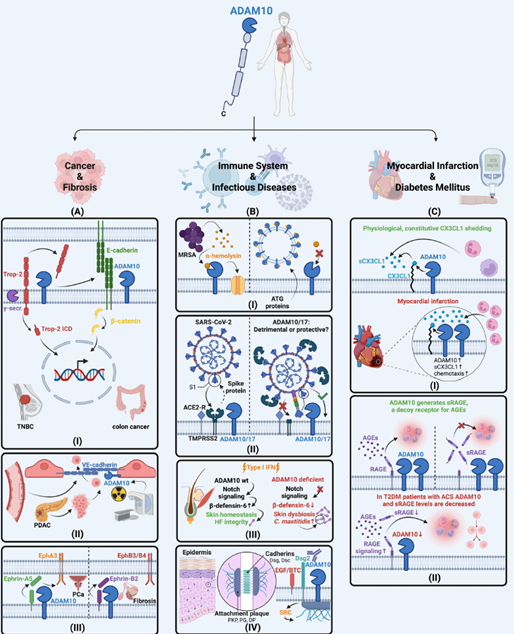
Involvement of ADAM10 in human diseases (Rosenbaum & Saftig, 2023, FEBS J.)
- Work in the lab contributed to a better understanding how tetraspanins, especially tetraspanin 15 and tetraspanin 3, modulate the cellular trafficking and activity of ADAM10.